rhAmpSeq™ CRISPR Library Kit
Our proprietary rhAmp™ PCR technology delivers optimal amplification with just two PCR reactions and minimal hands-on time. Combine the rhAmpSeq Library Kit with rhAmpSeq CRISPR Panels and rhAmpSeq Index Primers for rapid preparation of amplicon libraries for sequencing on Illumina® platforms.
NGS-ready amplicon libraries. Fast. Easy. Convenient.
Ordering
The rhAmpSeq CRISPR Library Kit comes with Analysis Credits for the rhAmpSeq CRISPR Data Analysis Tool, a cloud-based software package for quantification of CRISPR editing events. This tool generates publication-ready editing reports without the need for advanced bioinformatics skills.
- Sequencing-ready amplicon libraries for analyzing CRISPR on- and off-target editing results
- Low-input, uniform amplification for hundreds of target sites
- High coverage uniformity and high on-target rate
To complete your rhAmpSeq CRISPR Analysis System workflow, you will also need a rhAmpSeq CRISPR Panel and rhAmpSeq Index Primers.
Analysis Credits are provided with every rhAmpSeq CRISPR Library Kit purchased allowing you to use the rhAmpSeq CRISPR Analysis Data Tool. Each credit allows for the analysis of up to 500 targets for one, indexed sample. Upon purchase of this product, your activation code to redeem for Analysis Credits is emailed directly to you.
* This product is currently not available in China or Russia.
Product details
The rhAmpSeq CRISPR Library Kit contains two amplification mixes prepared for rapid, targeted, sequencing-ready amplicon libraries.
- rhAmpSeq Library Mix 1—formulated to reduce off-target amplification in multiplexed libraries. This mix is for Targeted rhAmp PCR 1 in the rhAmpSeq workflow (Figure 1).
- rhAmpSeq Library Mix 2—formulated for efficient amplification. This mix is for Indexing PCR 2 in the rhAmpSeq workflow. In this universal PCR step, rhAmpSeq Index Primers add index barcodes and Illumina P5/P7 adapter sequences to the library (Figure 1).
Harnessing the power of rhAmp PCR
Our proprietary rhAmp PCR technology drives the rhAmpSeq CRISPR Analysis System. This technology harnesses the intrinsic properties of the RNase H2 enzyme and RNA-base–containing blocked primers (rhAmp primers), minimizing primer dimers and enabling rhAmpSeq panels to be highly multiplexed.
Those two advantages—minimal primer dimers and high multiplexing capabilities—allowed us to develop the high-performance rhAmpSeq CRISPR Analysis System, whose fast, easy workflow requires only two PCR amplification steps (Figure 1) to generate amplicon libraries for Illumina platform sequencing.
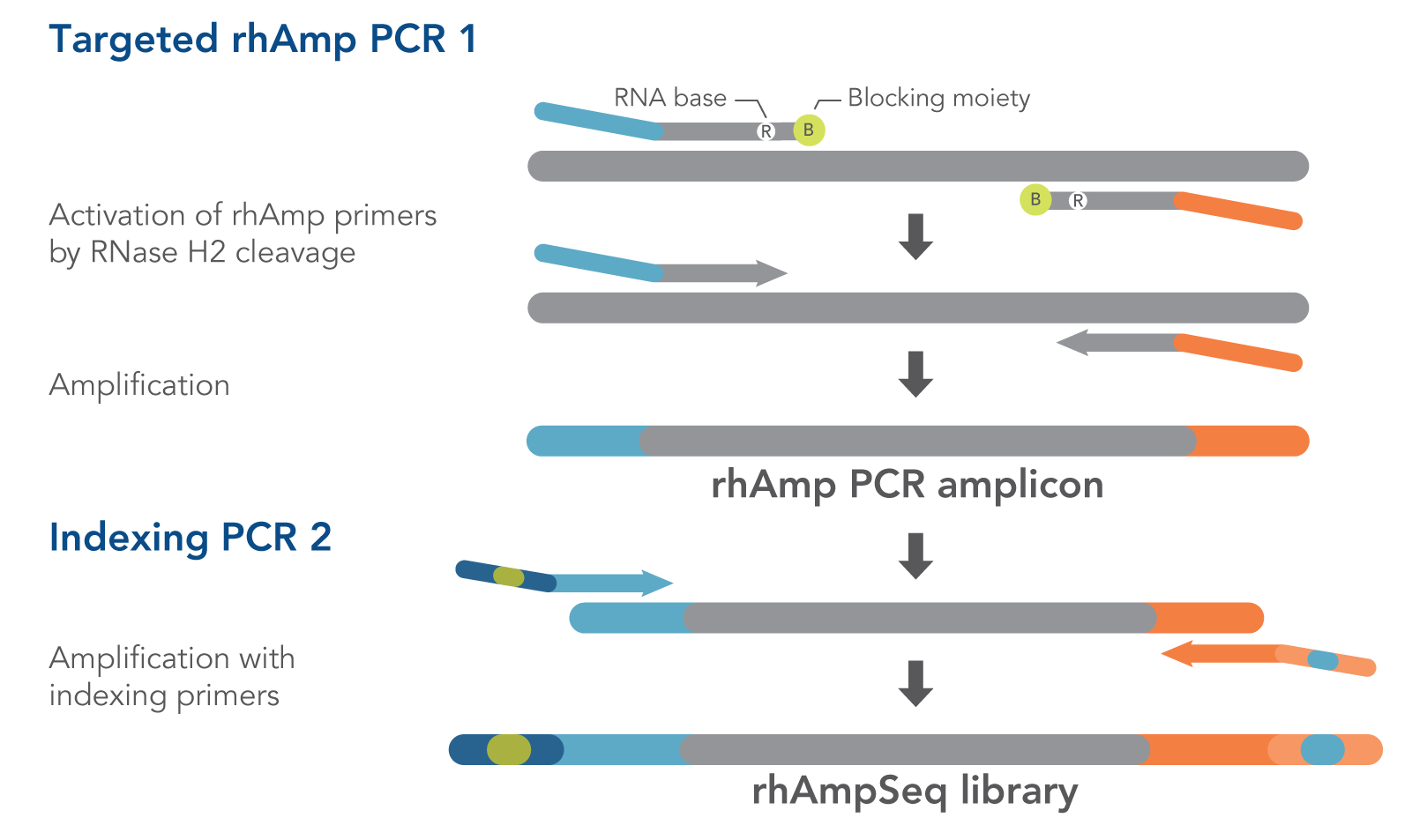
Figure 1. Details of amplification steps in the rhAmpSeq workflow. RNase H2 activates rhAmp primers by target-specific cleavage of the RNA base within the DNA:RNA duplex, removing a 3′ blocker. RNase H2 activity is mismatch sensitive, thus reducing the amount of amplification from non-specific hybridization and primer dimers. Only activated rhAmp primers can be extended to generate target amplicons.
Illumina sample barcodes and P5/P7 sequences are incorporated during Indexing PCR 2.
Technical details
Table 1. rhAmpSeq system features and specifications.
| Feature | Specification |
|---|---|
| Supported applications/protocols | On- and off-target analysis of genome editing experiments High-throughput analysis of on-target genome editing experiments |
| Insert size | Flexible (50–200 nt) |
| Custom panel size | Up to 5000 amplicons per panel |
| Sample indexing capability | 96 index sequences (up to 9216 combinations) |
| Compatible platforms | Illumina |
| Hands-on time* | 1–1.5 hr |
| Data analysis time** | 0.5–2 hr |
* Estimated time to process 12–96 samples using manual pipetting, including reaction setup, cleanup, library quantification, and normalization steps
** Estimated time to process sequencing data (up to 500 amplicons) through rhAmpSeq CRISPR Data
Analysis Tool
Product data
Effective on-target rate, uniform coverage with no primer-dimer amplification
The rhAmpSeq CRISPR Analysis System enables singleplex or multiplex analysis of CRISPR edits by next-generation sequencing (NGS) in less than a week. Our custom design tool ensures maximal compatibility between the target primers and the rhAmpSeq CRISPR Library Kit, preventing primer-dimer amplification during two simple PCR steps required for sequencing on any Illumina platform (Figure 2). The system includes access to an intuitive data analysis tool that generates publication-quality figures without needing advanced bioinformatics expertise.
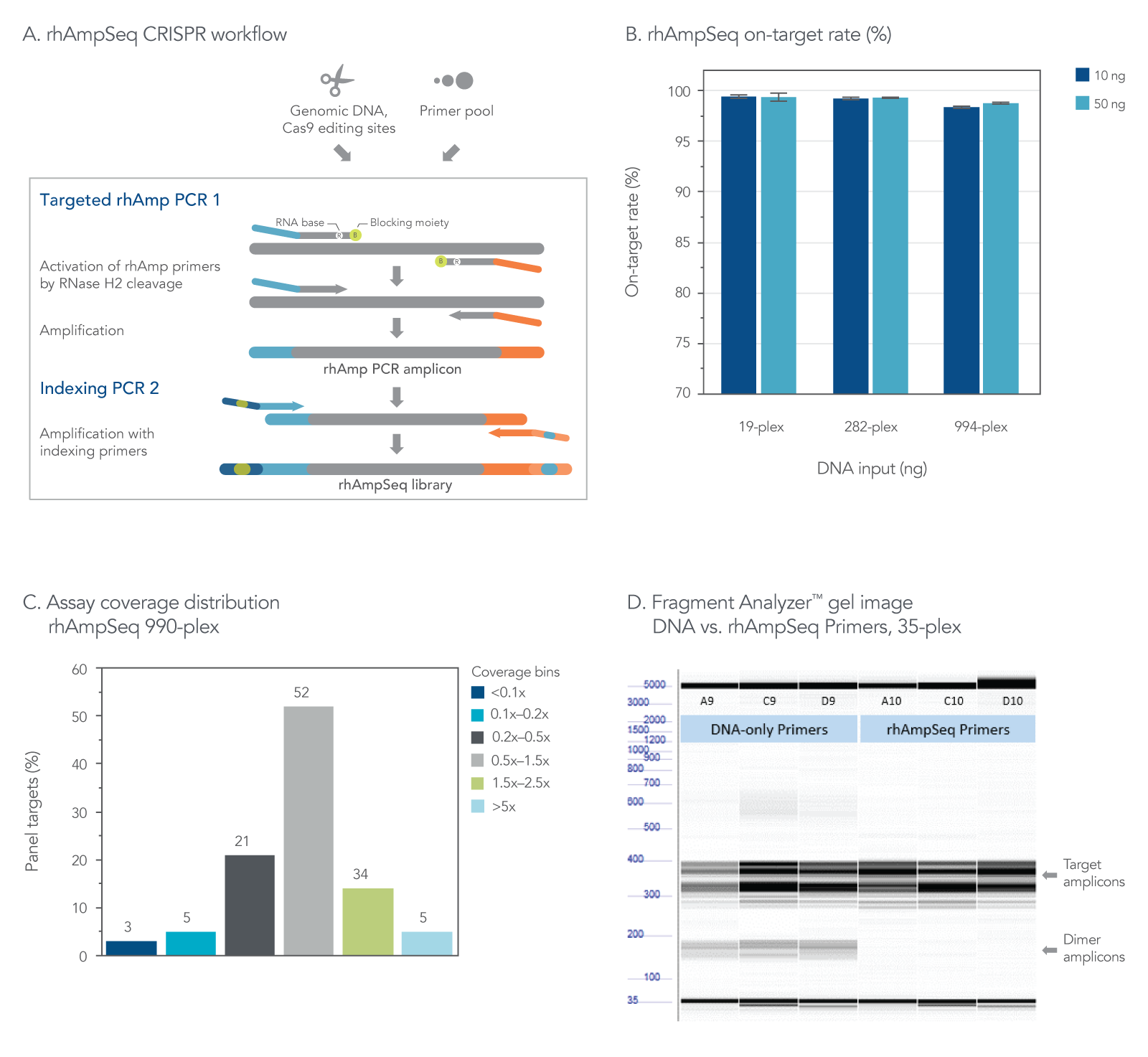
Figure 2. The rhAmpSeq CRISPR Analysis System provides efficient on-target rate and uniform coverage with no primer-dimer amplicons. (A) The workflow for targeted enrichment via amplification with the rhAmpSeq CRISPR Analysis System generates NGS-ready libraries in two straightforward PCR steps. (B) The rhAmpSeq CRISPR on-target rate represents the fraction of reads from expected targets in the rhAmpSeq CRISPR Panel relative to all mapped reads from the library. In this experiment, genomic DNA samples (Coriell) were amplified with a 19-, 282-, and 994-plex rhAmpSeq CRISPR Panel with 10 and 50 ng of input DNA. All panels were highly specific, giving on-target mapping rates of >98% (n = 3 amplifications per condition). (C) The uniformity of target amplification in a 990-plex is shown. In this experiment, 95% of all targets give coverage that is >0.2X of the mean coverage depth of the entire panel. (D) DNA-only primers compared to rhAmp primers show an accumulation of primer dimers in this 35-plex reaction with unblocked DNA primers, while no primer-dimers are observed when using our end-blocked, RNaseH2-cleavable, rhAmp primers.
Panel uniformity
High uniformity with rhAmpSeq CRISPR Panels allow for interrogation of on- and off-target editing in a single library prep with PCR amplification, as represented using the popular AAVS1 “safe harbor” site (Figure 3). All 28 empirically identified off-target sites can be verified when Cas9 is constitutively expressed in HEK293 cells. Using ribonucleoprotein (RNP) delivery of the Cas9/guide complex in conjunction with Alt-R™ S.p. HiFi Cas9 Nuclease V3 dramatically reduces off-target editing.
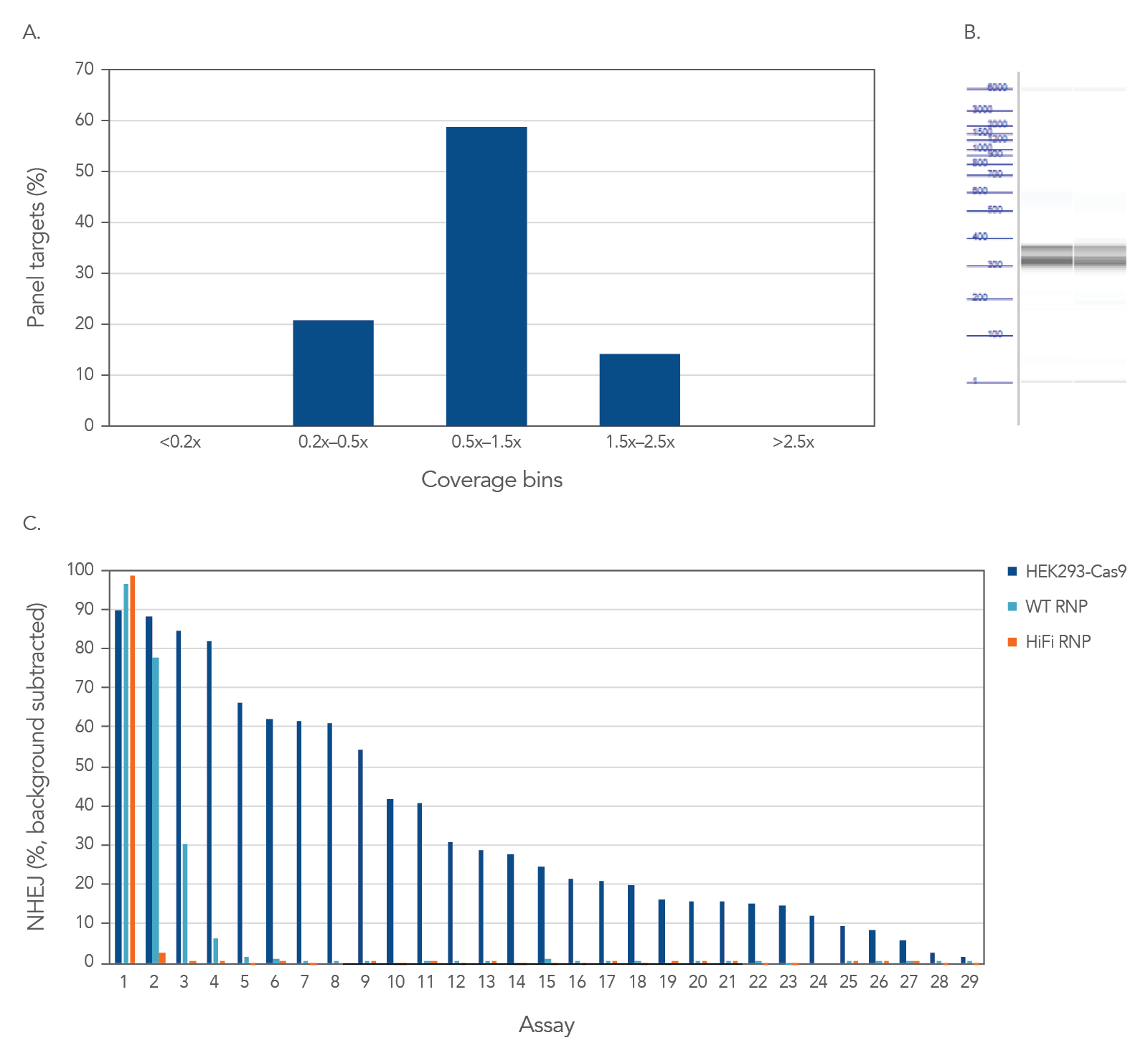
Figure 3. High uniformity with rhAmpSeq CRISPR Panels allow for interrogation of on- and off-target editing in a single library prep with PCR amplification. HEK293 cells constitutively expressing S.p. Cas9 nuclease were electroporated with 10 µM AAVS1-targeting Alt-R sgRNA. Alternatively, standard HEK293 cells were electroporated with 4 µM Alt-R wild-type (WT) or HiFi Cas9 Nuclease complexed to the AAVS1 sgRNA (at a 1:1.2 protein to gRNA ratio), including 4 µM Alt-R Cas9 Electroporation Enhancer using the Amaxa™ Nucleofector™ 96-well Shuttle™ System (Lonza) (n = 1 transfection per condition). gDNA was isolated and amplified using a custom rhAmpSeq CRISPR Panel containing amplicons for the on-target and 28 of the top off-target sites identified by GUIDE-Seq. Amplicon sequencing on the Illumina MiSeq (v2 chemistry, 150 bp paired-end reads) was performed and analyzed using the rhAmpSeq CRISPR Data Analysis Tool. (A) The histogram of panel coverage shows 100% of assays have read coverage depth that is >0.2X of the mean read coverage depth for all assays in the panel, indicating highly uniform enrichment via amplification. (B) Fragment analysis of final panels in duplicate shows the expected fragment sizes of 300–400 bp with no primer dimers present. (C) NHEJ editing for the on-target locus (Assay 1) and off-target loci (Assays 2-29) is reported using an untreated control for background subtraction. Background editing was <0.2% for all assays in this panel.
Batch analysis of single amplicon assays
While the rhAmpSeq CRISPR Analysis System is designed for multiplex analysis of on- and off-target editing, it also performs exceptionally well on batch analysis for single amplicon designs. Its analyses of on-target editing have high concordance with traditional workflows while offering a more simplified and cost-effective workflow that supports high-throughput screening applications (Figure 4).
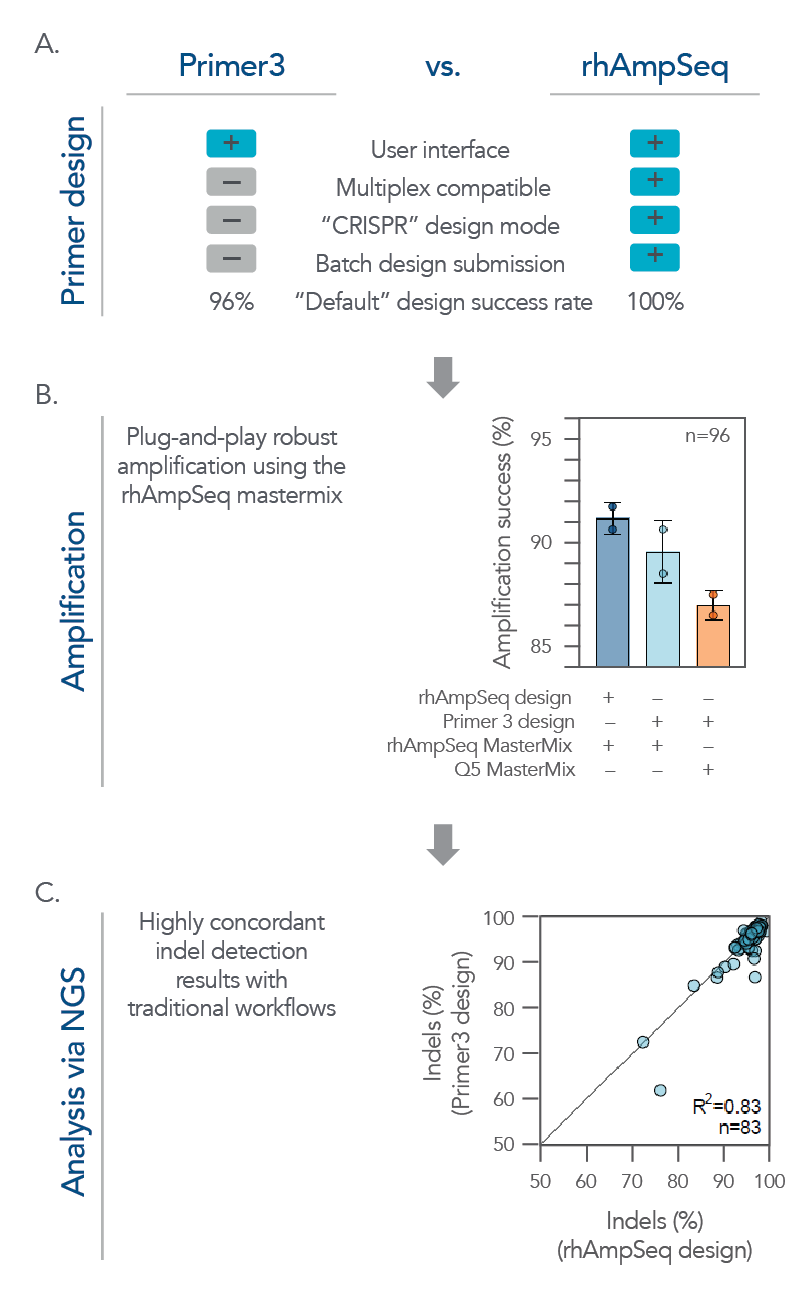
Figure 4. The rhAmpSeq CRISPR system supports a complete workflow for high-throughput, batch single amplicon design and analysis for on-target editing. (A) A batch of single amplicon assays (n = 96) were designed using the rhAmpSeq Design Tool or Primer3 (primers spaced >20 bp from the cut site with 130 bp design space) with default parameters, and the features/design success rates were compared. The rhAmpSeq Design Tool was more supportive than Primer3 to batch primer design. (B) Primers designed by Primer3 were synthesized as DNA-only primers, while primers designed by the rhAmpSeq Design Tool were synthesized as end-blocked, RNaseH2-cleavable rhAmp primers. DNA-only or rhAmp primers were used to amplify CRISPR-edited gDNA samples from HEK293 cells using either Q5® Hot Start High-Fidelity 2x Master Mix (NEB) or rhAmpSeq Master Mix. (C) NGS libraries from each combination of design/amplification were prepared and sequenced on an Illumina MiSeq system (2x150) and analyzed using the rhAmpSeq CRISPR Data Analysis Tool. Editing (% indels) concordance demonstrates that editing quantification between the two assay design strategies is equivalent.
Resources
Frequently asked questions
How long is my data retained in the rhAmpSeq CRISPR Data Analysis Tool?
The rhAmpSeq CRISPR Data Analysis Tool has retention policies for the following data types:
FASTQ files (input) = 30 days from last use
BED files (input) = 30 days if unpaired OR 5 years after pairing
All results files (output) = 1 year from date of generation
Please make sure these times are kept in mind for downloading/storing copies of results in longevity. It should be noted that there is no way to recover files after a retention period has lapsed. At this point the files will either need to be re-uploaded (inputs) or re-analyzed (outputs) by the end user to be available after the retention period has lapsed.
How sensitive is the rhAmpSeq™ CRISPR Analysis System?
With CRISPAltRations™, the software behind the rhAmpSeq CRISPR Data Analysis Tool, we analyzed the editing in three experimental replicates of 20 paired, treated, and control Cas9-edited loci. The treated samples had known indel editing levels (0.05, 0.10, 0.20, 0.50, 1.0, 2.0, and 3.4%), while the control samples had noise signals ranging from 0–0.4%.
Using the Fisher’s Exact Test (with a threshold of p<0.5), we show that 95% sensitivity and specificity were obtained for detecting indels down to 0.5%. This analysis assumed that there were:
- more than 5000 paired-end reads in both the treatment and control samples
- ·normal levels of background indel noise (<0.4% indels)
- treatment sample loci with editing >0.5%
- libraries prepared with the rhAmpSeq CRISPR Library Kit and sequencing data analyzed by the rhAmpSeq CRISPR Data Analysis Tool (powered by CRISPAltRations®).
Why is it that my main rhAmpSeq™ CRISPR Panel does not contain assays against all my targets?
The rhAmpSeq Design Tool aims to maximize PCR target specificity at intended loci and minimize any amplification of unintended loci that might consume valuable sequencing reads. Therefore, the design output can sometimes include not only the main panel, but also a secondary pool and possibly individual (singleton) primer sets requiring independent amplification.
Several factors can lead to more than one panel being designed:
- The PCR targets may be too close together. The rhAmpSeq Design Tool requires a minimum distance of 600 base pairs (bp) between targets. Targets that do not meet this minimum distance requirement impede having two unique primer pairs in a single pool. Typically, this results in an adjacent target needing to be covered by either a secondary pool, or as a singleton assay, if the assays cannot be merged.
- The algorithm may not be able to select a primer pair that is both specific enough to cover the PCR target of interest, and distinct enough to allow pooling (multiplexing) without creating primer dimers.
- The ideal primer pair for a given target may amplify a repetitive region. In this case, the optimal primer pair must remain a singleton primer pair to avoid impacting the operation of the primary pool.
- An assay with potential issues is less likely to be pooled due to the risk of impacting the overall panel results. Examples of these types of potential issues include the following:
- A primer overlaps common single nucleotide polymorphisms (SNPs) leading to potential inefficient amplification.
- The GC content of a primer or amplicon significantly differs from the panel’s average GC content.
- The Tm or secondary structure of the primers is not ideal.
- The amplicons contain homopolymers.
- The locus and design constraints do not permit the design of a primer pair that is expected to uniformly amplify with the assays in the primary pool.
What type of analysis can be performed using the rhAmpSeq™ CRISPR Data Analysis Tool?
The rhAmpSeq CRISPR Data Analysis Tool is capable of single-plex and multiplex analyses.
In singleplex analysis, the sample library has only one amplicon that is used to assess the editing of a single site in a sample. You would use singleplex analysis to assess numerous guide RNAs for on-target activity.
In multiplex analysis, the sample library is generated by multiplex PCR and is composed of multiple amplicons that assess the editing on multiple sites in a sample. You would use multiplex analysis to assess the on- and off-target editing of a guide RNA.
How do I design a rhAmpSeq™ CRISPR Panel to evaluate on- and off-target editing using the rhAmpSeq CRISPR Data Analysis Tool?
For compatibility with the rhAmpSeq CRISPR Data Analysis Tool, we recommend submitting the targets of interest to the rhAmpSeq Design Tool as a 6-column BED file that includes the following information: chromosome, start, stop, desired name, mismatch # (or a 0), and strand (+ or –).
Additionally, consider the following criteria with the BED file input:
- The BED file needs to be devoid of headers.
- The input to the tool should be the coordinates of your on- and off-target gRNA binding loci (i.e., usually 20–24 bases in length).
- The guides should include “strand” information. This is needed for the rhAmpSeq CRISPR Data Analysis Tool to report results accurately. Failing to provide strandedness will result in use of an incorrect, non-optimal editing window in the analysis software.
- Guide locations should NOT include the PAM sequence; remove the PAM sequence from the target location in the BED file. Failure to remove all the PAM information can adversely affect data analysis.
The rhAmpSeq CRISPR Data Analysis Tool has the following run restrictions:
- Multiplexed amplification pools are limited to 500 target sites per pool.
- Input FASTQ files should be <1 GB in file size.
Only the following organisms are supported by rhAmpSeq CRISPR Data Analysis Tool:
- Homo sapiens, human (hg38 & hg19)
- Mus musculus, mouse (mm10)
- Caenorhabditis elegans (Wbcel235)
- Rattus norvegicus, rat (rn6)
- Danio rerio, zebrafish (Grcz11)
- Macaca mulatta, rhesus monkey (rheMac8)
- Macaca fascicularis, crab-eating macaque (macFas5)
What is the Analysis Lab?
The Analysis Lab is a platform that IDT created to enable multiple researchers to collaborate and access the rhAmpSeq™ CRISPR Data Analysis Tool simultaneously, share experimental findings, and manage rhAmpSeq Analysis Credits.
Activation codes are redeemed, added to the platform, and then can be used to analyze data with the rhAmpSeq CRISPR Data Analysis Tool.
What is the analysis platform that powers the rhAmpSeq™ CRISPR Data Analysis Tool?
CRISPRAltRations is the analysis platform behind the rhAmpSeq CRISPR Data Analysis Tool.
This IDT-developed analysis pipeline is used for CRISPR on- and off-target editing analysis of next generation sequencing (NGS) data generated from amplicon sequencing. The analysis platform available through the rhAmpSeq CRISPR Data Analysis Tool utilizes cloud-hosted computational resources for data processing.
Briefly, the CRISPR workflow is as follows:
1.
Read pairs are identified and merged from paired-end sequencing.
2. Reads are binned to the expected amplicons, resulting from targeted amplification library preparation (e.g., the rhAmpSeq CRISPR Library Kit).
3. A Cas-enzyme
specific aligner aligns the read to the expected amplicon.
4. Variants are called and summarized.
Although this workflow is relatively common to most software tools that analyze NGS data derived from CRISPR screens, the rhAmpSeq CRISPR Data Analysis Tool has key improvements that help to enable more correct variant identification, including: a Cas-specific aligner; an optimized default variant identification window; systematically substantiated program parameters that use open source tools to provide high-quality results.
How do I find my rhAmpSeq Analysis Credit balance?
The rhAmpSeq Analysis Credits balance can be found in your Analysis Lab, as well as on the rhAmpSeq™ CRISPR Data Analysis Tool.
Additional information, such as the expiration dates for your Analysis Credits, is listed in the Analysis Credit balance details in the Analysis Lab.
Do IDT's rhAmpSeq™ Analysis Credits expire?
Yes, your rhAmpSeq Analysis Credits expire after 1 year.
IDT's rhAmpSeq Analysis Credits are automatically redeemed from the earliest expiration date to the latest.
View the expiration dates on your rhAmpSeq Analysis Credits in the Analysis Credit balance details window found in the Analysis Lab.
How do I analyze CRISPR-editing next generation sequencing (NGS) data with the rhAmpSeq™ CRISPR Data Analysis Tool?
For compatibility with the rhAmpSeq CRISPR Data Analysis Tool, we recommend submitting the targets of interest to the rhAmpSeq Design Tool as a 6-column BED file that includes the following information: chromosome, start, stop, desired name, mismatch # (or a 0), and strand (+ or –).
You can use the rhAmpSeq CRISPR Data Analysis Tool user guide for further information about input file formatting.