xGen™ SARS-CoV-2 Amplicon Panels
Targeted sequencing to research SARS-CoV-2 viral genomics
Overlapping primers produce 99.7%† genomic coverage of SARS-CoV-2 lineages. These consistent primers obtain sequence data from viral concentrations as low as 10–100 viral copies without the need to evaluate new primer sets for emerging lineages.
xGen NGS—made for COVID-19 research.
Ordering
The xGen SARS-CoV-2 Amplicon Panels enable identification of SARS-CoV-2 variants from research samples such as nasopharyngeal/oropharyngeal swabs, sputa, stool, and wastewater. The xGen ACE2 Gene Amplicon Panel amplifies the angiotensin converting enzyme 2 gene which has been proven to be the SARS-CoV-2 spike protein receptor [1]. The xGen SARS-CoV-2 Sgene panel amplifies the viral gene for the spike protein only.
The xGen SARS-CoV-2 Amplicon Panels support:
- 99.7% genomic coverage†
- Full genomic coverage of lineages from a single primer set
- Sequence data from viral titers as low as 10–100 viral copies
- cDNA-to-sequencer in 3 hours
- Up to 1536 UDIs
† See Product Data section for more information.
Transform Your NGS Workflow with Automation
Looking to streamline your NGS workflows? Discover how automation can enhance efficiency and consistency in your lab with our NGS Automation solutions.
Request a consultation
Want to learn more about our xGen™ Amplicon Panels—developed with super amplicon technology—and how to customize your panel or spike-in genes in our predesigned panels? Your time is valuable—we’ll prioritize your inquiry and be in touch to discuss it ASAP.
Request a consultationRush University Medical Center
Product details
xGen SARS-CoV-2 Amplicon Panels, previously known as Swift Normalase™ Amplicon Panel (SNAP) SARS-CoV-2 enables researchers to identify SARS-CoV-2 strains, including variants, by creating next generation sequencing (NGS) libraries. The sequencing data can then answer research questions, such as:
- Which viral strains account for most infections within a local population?
- What new strains are emerging?
IDT’s predesigned amplicon panels for COVID research can aid in identifying the extent and pattern of viral spread within a population and allow researchers to quantify the effectiveness of intervention measures.
The xGen SARS-CoV-2 Amplicon Panel enables identification of SARS-CoV-2 variants from a wide array of research sample types such as nasopharyngeal/oropharyngeal swabs, sputa, stool, and wastewater. This panel covers 99.7% of the SARS-CoV-2 genome for identification of mutations from Alpha and Delta strains (see Product Data).
The xGen SARS-CoV-2 Sgene Amplicon Panel provides 100% coverage of the gene which encodes for the spike protein in the virus that causes COVID-19. Identify the presence of emerging viral strains which contain mutations in the Sgene and model potential changes in transmission patterns based upon those mutations.
The xGen ACE2 Gene Amplicon Panel contains 41 amplicons with an average size of 150 bp that provides comprehensive coverage of all coding regions of ACE2. Sequencing ACE2 has the potential to provide insight into disease outcome and facilitate further epidemiological investigations.
cDNA-to-sequencer in 3 hours
To sequence SARS-CoV-2 viral genomic material, first prepare first- or second-strand cDNA from samples such as nasopharyngeal/oropharyngeal swabs, sputa, stool, or wastewater (Figure 1). Generate an NGS library using tiled primer pairs in a single tube to target the 29.9 kb viral genome. Primers were designed against the NCBI Reference Sequence NC_045512.2 (severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1, complete genome) (Table 1). Following a positive result by qPCR, use excess cDNA to perform variant calling and identify the viral strain.
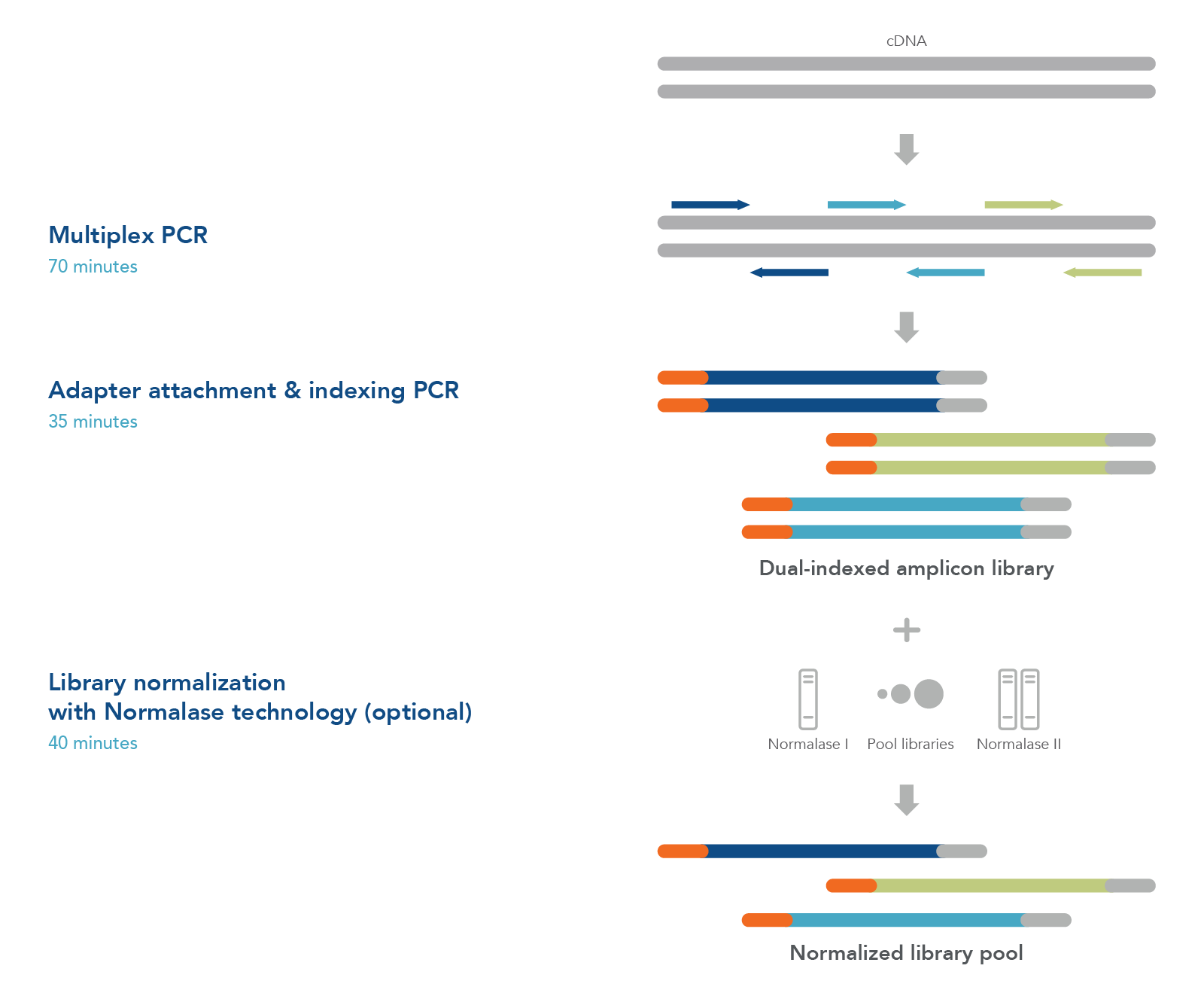
Figure 1. One-tube workflow prepares normalized libraries from cDNA in 3 hours by replacing qPCR library quantification and normalization with the proprietary Normalase™ technology (included in the kit).
Table 1. NGS for SARS-CoV-2 specifications.
| Features | Specifications |
|---|---|
| Design coverage and panel information |
|
| Input material |
|
| Time | 2 hours cDNA-to-Library |
| 3 hours cDNA-to-Normalized-Library Pool | |
| Multiplexing capability |
|
| Compatible with other indexes? | Yes |
| Recommended depth |
|
Product data
99.7% SARS-CoV-2 genomic coverage
The xGen SARS-CoV-2 Amplicon Panel uses overlapping primers to generate 345 amplicons, sized 116–255 bp (average 150 bp), along the length of the 29.9 kb viral genome, obtaining 99.7% coverage of the genome (Figure 2). Overlapping primers ensure that variants are identified, even when the mutation interferes with a primer binding site. Comprehensive mutation identification is crucial for tracking nucleotide variants and improve understanding of virus evolution, transmission, and pathogenesis.
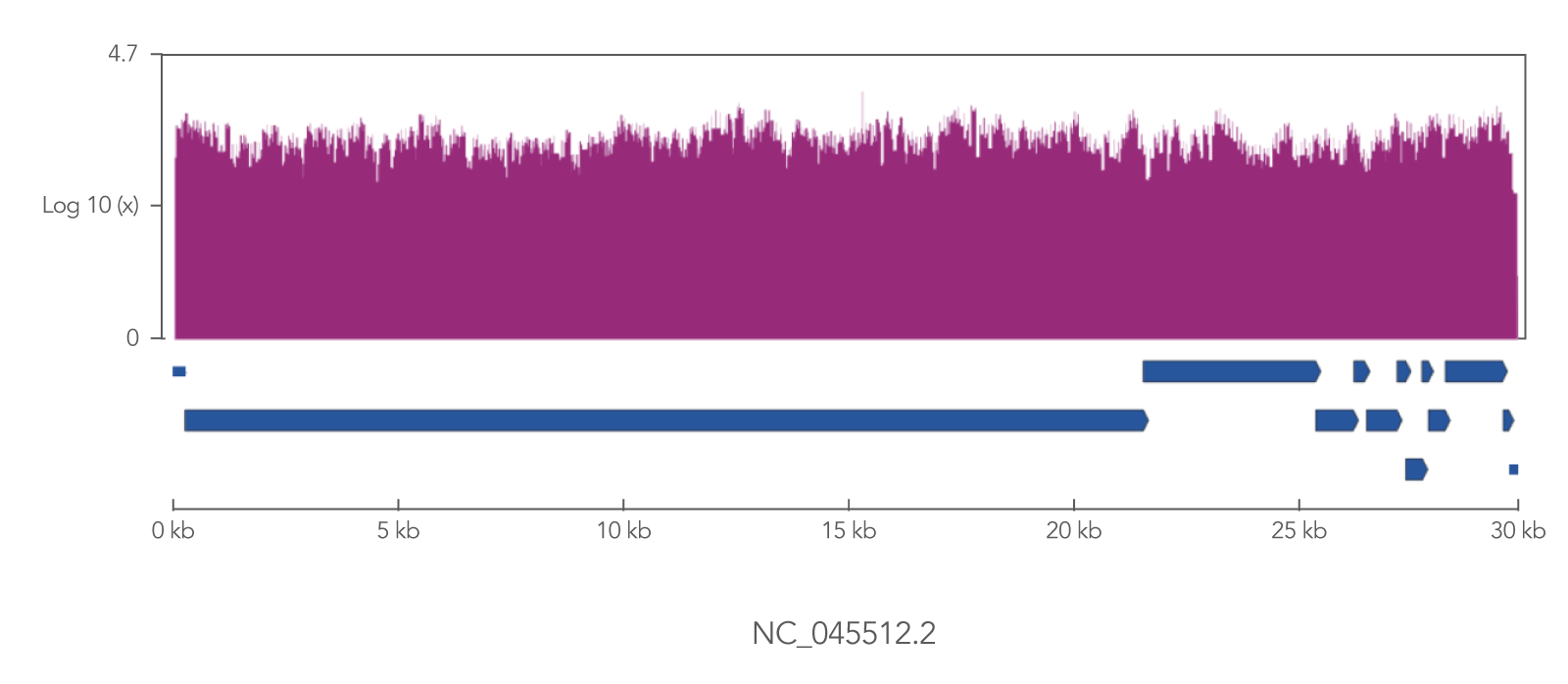
Figure 2. The xGen SARS-CoV-2 Amplicon Panel provides 99.7% coverage of the SARS-CoV-2 genome. RNA was isolated from gamma-irradiated and sonicated cell lysate from Vero E6 cultured monkey kidney epithelial cells (ATCC® CRL-1586™) infected with SARS-CoV-2 isolate USA-WA1/2020 (BEI Resources, Cat. No. NR-52287). 100,000 copies of SARS-CoV-2 RNA was converted into cDNA using the SuperScript™ IV Kit (Thermo Fisher Scientific). The resulting NGS library generated with the xGen SARS-CoV-2 Amplicon Panel was sequenced 2 x 150 bp on a MiniSeq® System (Illumina). Resulting reads were downsampled to 280k reads per sample (n = 2) for analysis. Genomic coverage for a representative sample is shown.
Full genomic coverage of lineages with a single primer set
Using a single, consistent primer set, xGen SARS-CoV-2 Amplicon Panel has been shown to cover multiple lineages, including sub-lineages of Omicron (Figure 3), Delta, and Alpha (Figure 4). The primer set has remained unchanged since its release in 2020, preventing the need for frequent changes to primer designs.
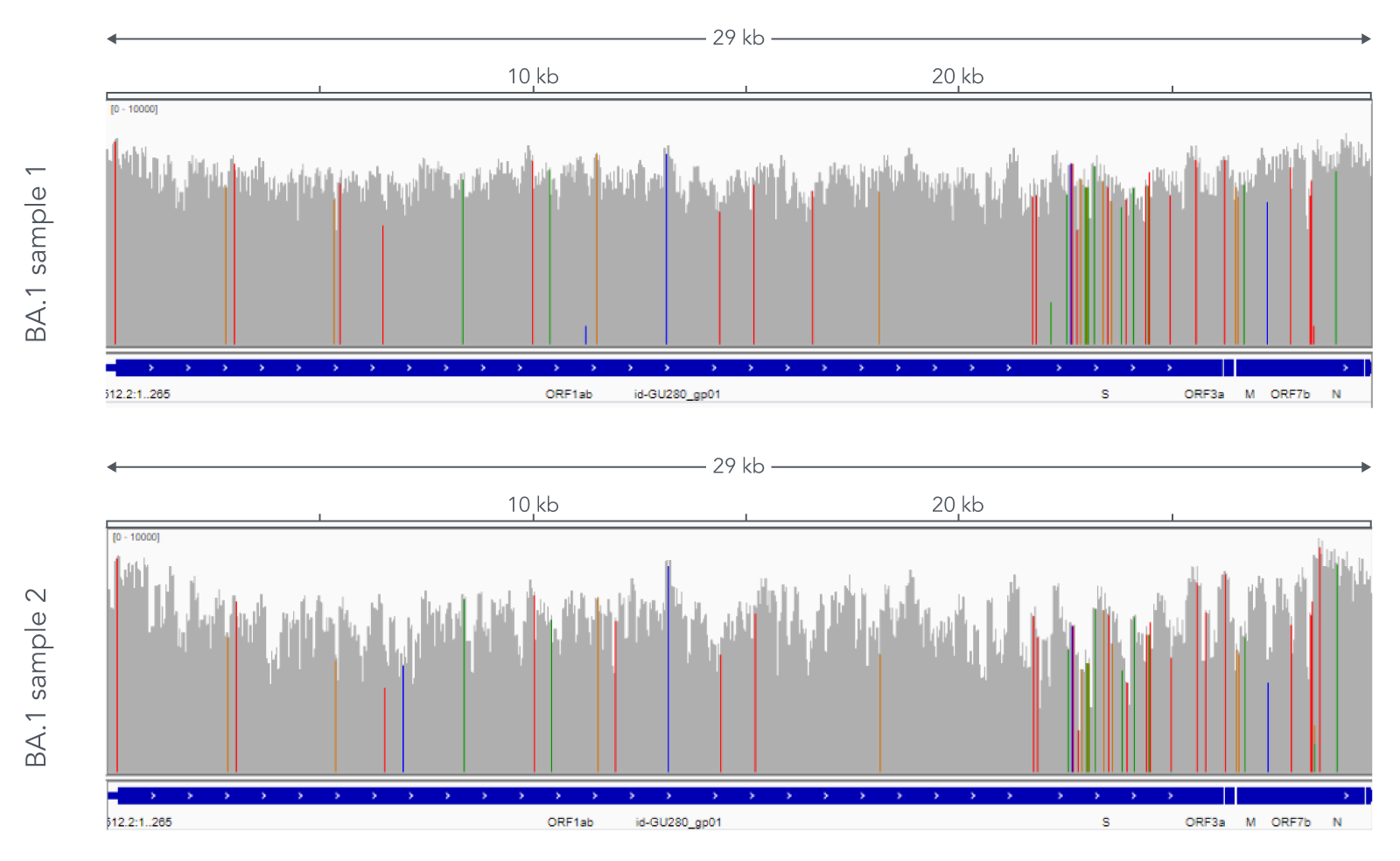
Figure 3. Two representative examples of sequence coverage obtained from Omicron SARS-CoV-2 RNA. RNA was extracted from nasopharyngeal swabs and converted into cDNA using the SuperScript IV Kit (Thermo Fisher Scientific). The resulting library generated with the xGen SARS-CoV-2 Amplicon Panel was sequenced 2 x 150 bp on a MiniSeq System (Illumina). Resulting reads were downsampled to 83,000 reads per sample for analysis. Genomic coverage for two BA.1 lineages is shown. Ct values are 13.6 and 19.9. A total of 93 biological research samples that exhibited S gene dropout by qPCR were analyzed, 90 of which were identified as Omicron variants.
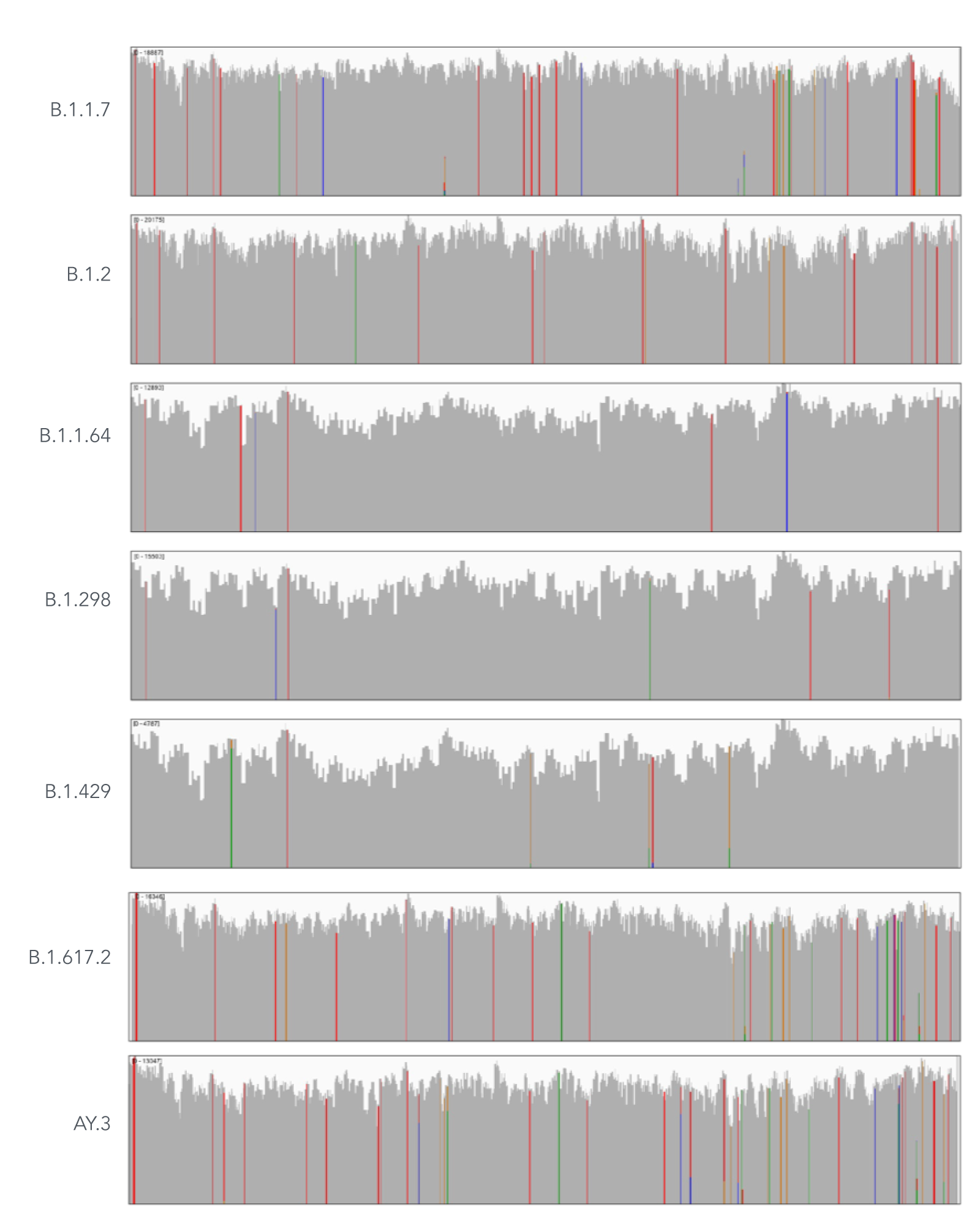
Figure 4. An example of coverages obtained from B.1.1.7 (Alpha), B.1.2, B.1.1.64, B.1.298, B.1.429 (Epsilon), B.1.617.2 (Delta), and AY.3 (Delta) SARS-CoV-2. RNA was extracted from nasopharyngeal or oropharyngeal swabs and converted into cDNA using the SuperScript IV Kit (Thermo Fisher Scientific). The resulting library generated with the xGen SARS-CoV-2 Amplicon Panel was sequenced 2 x 150 bp on a MiniSeq System (Illumina). Resulting reads were downsampled to 83,000 reads per sample for analysis. Genomic coverage for a representative sample per lineage is shown.
Research Investigation
Case Study I Genemarkers, LLC
Genemarkers, LLC, and Swift Biosciences collaborated to identify mutations and variants from SARS-CoV-2 samples sourced in Kalamazoo, Michigan. Samples were anticipated to be U.K. variants (B.1.1.7) due to the observation of S gene dropout in PCR testing. After generating an NGS library with the xGen SARS-CoV-2 Amplicon Panel and sequencing, the mutation pattern of SARS-CoV-2 B.1.1.7 was not observed in one sample, leading to clarification on variants present in the Kalamazoo area.
Obtain genomes from viral titers as low as 10–100 viral copies
10 to 1 million viral genome copies (Figure 5) are sufficient to generate NGS libraries using the xGen SARS-CoV-2 Amplicon Panel. Mixed RNA samples were converted into first strand cDNA and used to create sequencing libraries with the xGen SARS-CoV-2 Amplicon Panel. Libraries were enzymatically normalized to 4 nM using the Normalase workflow.
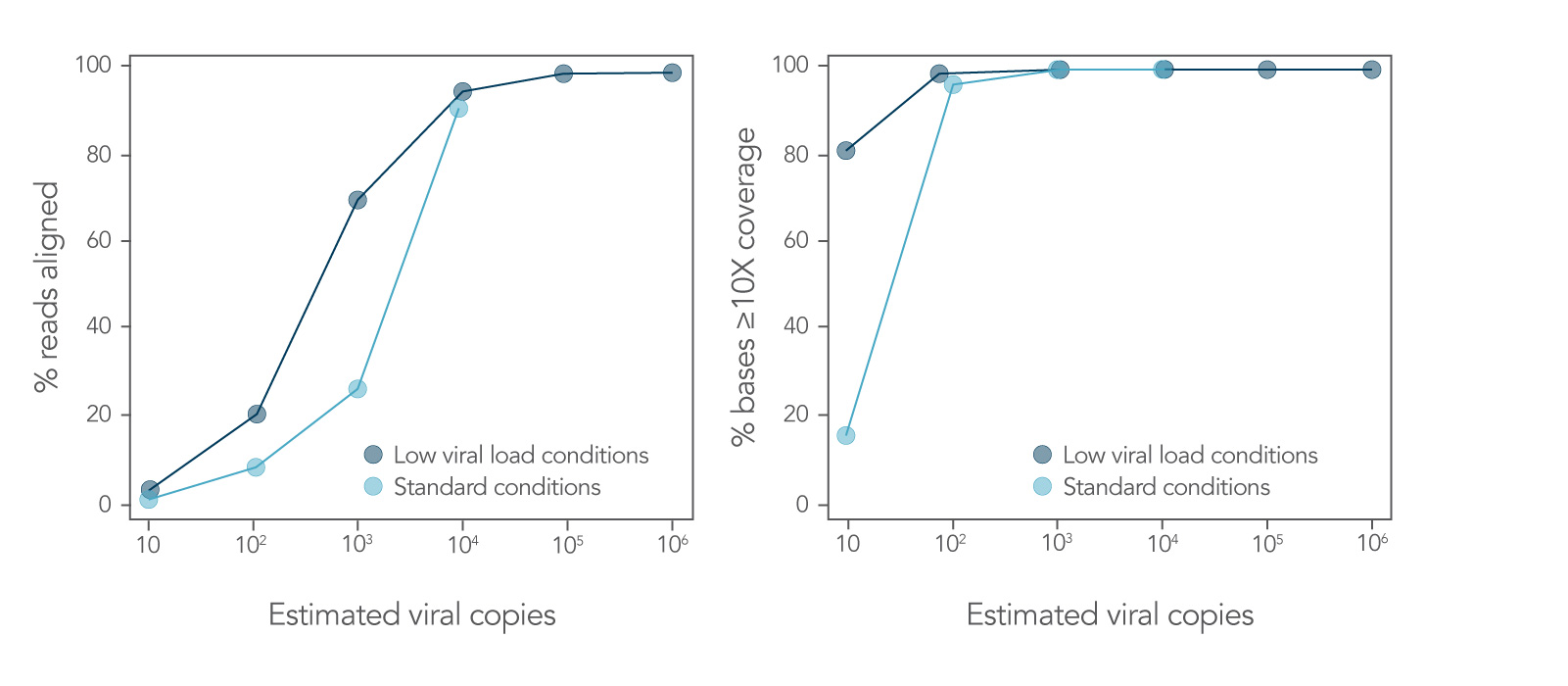
Figure 5. Obtain genomes from as few as 10–100 viral copies. SARS-CoV-2 RNA USA-WA1/2020 (BEI Resources, catalog # NR-52287) was mixed with Universal Human Reference RNA (Agilent, catalog # 740000) and converted into first-strand cDNA using the Superscript® IV First-Strand Synthesis System (Thermo Fisher Scientific). The resulting library generated with the xGen SARS-CoV-2 Amplicon Panel was sequenced on a MiniSeq System (Illumina) at 2 x 150 bp. Resulting reads were downsampled to 280k reads per sample (n = 2). Genomic copies identified are shown.
Resources
Frequently asked questions
Do I have to use the Normalase™ Module to finish my xGen™ Amplicon libraries?
No.
After the Indexing PCR step and the final bead-based size selection/cleanup, xGen Amplicon libraries may be quantified with conventional methods such as Qubit™, Bioanalyzer™ (Aligent), or qPCR and normalized by manual pooling.
Alternatively, they can be normalized enzymatically with the included xGen Normalase reagents.
Can I use my own UDI primers in the xGen™ Amplicon Sequencing workflow?
Yes.
Please contact Scientific Application Support team if you would like assistance confirming compatibility of your own primers with the xGen Amplicon Sequencing workflow.
Note that using your own UDI primers without Normalase™ modifications will make the amplicon libraries incompatible with the downstream Normalase workflow.
What reverse transcription (RT) module is recommended?
The xGen™ SARS-CoV-2 Amplicon Panel used with the xGen Amplicon Core Kit and indexing primers supports first- or second-strand cDNA, generated from the SARS-CoV-2 RNA genome, as input.
Select a cDNA synthesis module that supports two-step RT-PCR, includes random primers, and has a processivity of >1 kb.
The panel has been tested with Superscript® IV First-Strand Synthesis System (Thermo Fisher Cat. No. 18091050). The manufacturer’s protocol was followed as written using random primers and associated specifications. Steps involving the optional host gDNA/RNA removal and RNAse H were not performed.
How do I analyze sequencing data from libraries generated with the xGen™ SARS-CoV-2 Amplicon Panel?
For customers using the xGen™ SARS-CoV-2 Amplicon Panel who are comfortable with command line tools, and who want a ready-to-use variant calling analysis workflow that they can run on a local Linux machine, IDT offers a full variant calling workflow with all the tools and reference files pre-installed and configured in a Docker image available for download. See the xGen SARS-CoV-2 Amplicon Panel dockerized data analysis guidelines Tech Note for this information.
For details about primer trimming, review the Primerclip-A Tool for Trimming Primer Sequences App Note.
How do I convert qRT-PCR Ct values to SARS-CoV-2 viral copies?
A Ct value obtained from qRT-PCR can be converted to viral copy number if a standard/control of known copy number was also used in the assay.
If no standard was used, the Ct value can only give an approximate magnitude of starting viral copies. This is because Ct values can vary depending on the protocol/reagents being used as well as on which part of the SARS-CoV-2 genome is being targeted.
Although an exact copy number cannot be calculated, it is generally considered that a Ct value ≥35 represents a very low viral titer (<100 genome copies).
The xGen™ SARS-CoV-2 Amplicon Panel used with the xGen Amplicon Core Kit and indexing primers supports a wide range of inputs/Ct values. A low input protocol has been provided for samples with low viral titer. We recommend using the low input protocol for Ct >28.
References
- Yan R, Zhang Y, Li Y, et al. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science (New York, NY). 2020;367(6485):1444-1448.